Functional analysis: PROGENy / DoRothEA / CytoSig#
To facilitate the interpretation of scRNA-seq gene expression readouts and their differences across conditions we can summarize the information and infer pathway activities from prior knowledge.
In this notebook we use decoupler [Badia-I-Mompel et al., 2022], a tool that contains different statistical methods to extract biological activities from omics data using prior knowledge.
We run the method on the results from the differential gene expression analysis that compared scRNAseq data from LUAD and LUSC using celltype specific
pseudo-bulk data.
Note
Curated gene sets are readily available from MSigDB [Liberzon et al., 2015] and include amongst others gene ontology (GO)-terms [Consortium, 2004], KEGG [Kanehisa, 2000] pathways and Reactome [Gillespie et al., 2021] pathways. In addition, several recent efforts focus on the curation of high-quality gene signatures: PROGENy [Schubert et al., 2018] provides a set of reliable signatures for 14 cancer pathways derived from hundreds of perturbation experiments. Unlike signatures based on genes directly involved in a pathway (e.g. from KEGG), these signatures consist of downstream “pathway-responsive genes” that are differentially expressed when the pathway is perturbed (figure 1.10). This is beneficial as pathways tend to be regulated via post-translational modifications. Naturally, these modifications cannot be measured using RNA-sequencing, but requires either phosphoproteomics or targeted antibody assays. CytoSig [Jiang et al., 2021] is a similar effort focussing on cytokine signaling signatures. Based on more than 2000 cytokine treatment experiments, the authors derived 52 signatures. DoRothEA [Garcia-Alonso et al., 2019] integrates transcription factor activity signatures from manually curated resources, large-scale experiments
We infer activities of the following types:
Pathways from PROGENy [Schubert et al., 2018]
Transcription factors from DoRothEA [Garcia-Alonso et al., 2019]
Molecular Signatures (MSigDB) [Liberzon et al., 2015]
Cytokine signaling (CytoSig) [Jiang et al., 2021]
%load_ext autoreload
%autoreload 2
import os
import re
import urllib.request
from pathlib import Path
import decoupler as dc
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import scanpy as sc
import seaborn as sns
import statsmodels.stats.multitest
from IPython.display import display
import atlas_protocol_scripts as aps
1. Configure paths#
Define paths to input files
adata_path: Path to anndata filedeseq_path: Path to directory where DESeq2 differential expression results are stored
adata_path = "../../data/input_data_zenodo/atlas-integrated-annotated.h5ad"
deseq_path = "../../data/results/differential_expression"
Define and create output directory
results_dir = "../../data/results/10_functional_analysis"
os.makedirs(results_dir, mode=0o750, exist_ok=True)
Define paths to network/model files. These will be automatically downloaded and stored in these files in a later step.
tfnet_file = Path(results_dir, "tf_net_dorothea_hs.tsv")
pwnet_file = Path(results_dir, "pw_net_progeny_hs.tsv")
msigdb_file = Path(results_dir, "msigdb_hs.tsv")
cytosig_file = Path(results_dir, "cytosig_signature.tsv")
2. Load data#
Load AnnData object
adata = sc.read_h5ad(adata_path)
print(f"Anndata has: {adata.shape[0]} cells and {adata.shape[1]} genes")
Anndata has: 62119 cells and 17837 genes
Retrieve and load DoRothEA data. We limit the analysis to the three highest confidence levels A, B and C.
if tfnet_file.exists():
tfnet = pd.read_csv(tfnet_file, sep="\t")
else:
tfnet = dc.get_dorothea(organism="human", levels=["A", "B", "C"])
tfnet.to_csv(tfnet_file, sep="\t", index=False)
Retrieve and load PROGENy data
# Retrieve Progeny db
if pwnet_file.exists():
pwnet = pd.read_csv(pwnet_file, sep="\t")
else:
pwnet = dc.get_progeny(organism="human", top=100)
pwnet.to_csv(pwnet_file, sep="\t", index=False)
Retrieve and load data from MSigDB. Here, we filter for the “hallmark” gene sets.
# Retrieve MSigDB resource
if msigdb_file.exists():
msigdb = pd.read_csv(msigdb_file, sep="\t")
else:
msigdb = dc.get_resource("MSigDB")
# Filter by a desired geneset collection, for example hallmarks
msigdb = msigdb[msigdb["collection"] == "hallmark"]
msigdb = msigdb.drop_duplicates(["geneset", "genesymbol"])
msigdb.to_csv(msigdb_file, sep="\t", index=False)
Retrieve and load the CytoSig signatures
# Retrieve CytoSig signature
if cytosig_file.exists():
cytosig_signature = pd.read_csv(cytosig_file, sep="\t")
else:
urllib.request.urlretrieve(
"https://github.com/data2intelligence/CytoSig/raw/master/CytoSig/signature.centroid.expand",
cytosig_file,
)
cytosig_signature = pd.read_csv(cytosig_file, sep="\t")
3. Define contrasts#
Here we define the gene expression contrasts/comparisons for which we to run the functional analyses.
contrasts is a list of dicts with the following keys:
name: contrast namecondition: test groupreference: reference group
For this tutorial, we only show one single comparison: LUAD vs. LUSC.
contrasts = [
{"name": "LUAD_vs_LUSC", "condition": "LUAD", "reference": "LUSC"},
]
contrasts
[{'name': 'LUAD_vs_LUSC', 'condition': 'LUAD', 'reference': 'LUSC'}]
Create result directories for each contrast
for contrast in contrasts:
res_dir = Path(results_dir, contrast["name"].replace(" ", "_"))
os.makedirs(res_dir, mode=0o750, exist_ok=True)
contrast["res_dir"] = res_dir
Define column in
adata.obsthat contains the desired cell-type annotation. This must match the column used in the Differential gene expression section.
cell_type_class = "cell_type_coarse"
Make list of all cell types in the specified column
cell_types = adata.obs[cell_type_class].unique()
print(f"Cell types in {cell_type_class} annotation:\n")
for ct in cell_types:
print(ct)
Cell types in cell_type_coarse annotation:
B cell
T cell
Epithelial cell
Macrophage/Monocyte
Mast cell
Plasma cell
cDC
Stromal
NK cell
Endothelial cell
pDC
Neutrophils
4. Read DESeq2 results#
Load TSV files created in the Differential gene expression step.
for contrast in contrasts:
print(f"Working on: {contrast['name']}")
de_res = {}
for ct in cell_types:
ct_fs = re.sub("[^0-9a-zA-Z]+", "_", ct)
deseq_file = Path(deseq_path, contrast["name"], ct_fs, ct_fs + "_DESeq2_result.tsv")
if os.path.exists(deseq_file):
print(f"Reading DESeq2 result for {ct}: {deseq_file}")
de_res[ct] = pd.read_csv(deseq_file, sep="\t").assign(cell_type=ct)
else:
print(f"No DESeq2 result found for: {ct}")
contrast["cell_types"] = de_res.keys()
contrast["de_res"] = de_res
Working on: LUAD_vs_LUSC
Reading DESeq2 result for B cell: ../../data/results/differential_expression/LUAD_vs_LUSC/B_cell/B_cell_DESeq2_result.tsv
Reading DESeq2 result for T cell: ../../data/results/differential_expression/LUAD_vs_LUSC/T_cell/T_cell_DESeq2_result.tsv
Reading DESeq2 result for Epithelial cell: ../../data/results/differential_expression/LUAD_vs_LUSC/Epithelial_cell/Epithelial_cell_DESeq2_result.tsv
Reading DESeq2 result for Macrophage/Monocyte: ../../data/results/differential_expression/LUAD_vs_LUSC/Macrophage_Monocyte/Macrophage_Monocyte_DESeq2_result.tsv
Reading DESeq2 result for Mast cell: ../../data/results/differential_expression/LUAD_vs_LUSC/Mast_cell/Mast_cell_DESeq2_result.tsv
Reading DESeq2 result for Plasma cell: ../../data/results/differential_expression/LUAD_vs_LUSC/Plasma_cell/Plasma_cell_DESeq2_result.tsv
Reading DESeq2 result for cDC: ../../data/results/differential_expression/LUAD_vs_LUSC/cDC/cDC_DESeq2_result.tsv
Reading DESeq2 result for Stromal: ../../data/results/differential_expression/LUAD_vs_LUSC/Stromal/Stromal_DESeq2_result.tsv
Reading DESeq2 result for NK cell: ../../data/results/differential_expression/LUAD_vs_LUSC/NK_cell/NK_cell_DESeq2_result.tsv
Reading DESeq2 result for Endothelial cell: ../../data/results/differential_expression/LUAD_vs_LUSC/Endothelial_cell/Endothelial_cell_DESeq2_result.tsv
No DESeq2 result found for: pDC
Reading DESeq2 result for Neutrophils: ../../data/results/differential_expression/LUAD_vs_LUSC/Neutrophils/Neutrophils_DESeq2_result.tsv
Convert DE results to p-value/logFC/stat matrices as used by decoupler
# Concat and build the stat matrix
for contrast in contrasts:
lfc_mat, fdr_mat = aps.tl.long_form_df_to_decoupler(pd.concat(contrast["de_res"].values()), p_col="padj")
stat_mat, _ = aps.tl.long_form_df_to_decoupler(
pd.concat(contrast["de_res"].values()), log_fc_col="stat", p_col="padj"
)
contrast["lfc_mat"] = lfc_mat
contrast["stat_mat"] = stat_mat
contrast["fdr_mat"] = fdr_mat
display(lfc_mat)
display(fdr_mat)
display(stat_mat)
| gene_id | A1BG | A1BG-AS1 | A2M | A2M-AS1 | A2ML1 | A4GALT | A4GNT | AAAS | AACS | AADAC | ... | ZW10 | ZWILCH | ZWINT | ZXDA | ZXDB | ZXDC | ZYG11A | ZYG11B | ZYX | ZZEF1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cell_type | |||||||||||||||||||||
| B cell | -0.363007 | -0.906790 | -0.934158 | 0.000000 | 0.000000 | 1.306582 | 0.000000 | 0.783459 | 0.511908 | 0.000000 | ... | 1.573657 | -0.915355 | 3.137976 | -0.539899 | -0.685941 | -0.482270 | -1.651918 | 1.164252 | -0.682958 | -0.776318 |
| Endothelial cell | 0.833616 | 1.614074 | -0.318141 | -2.257129 | 0.000000 | -1.719525 | 0.000000 | -3.869248 | -1.668956 | -4.995006 | ... | -0.893945 | -4.927789 | -0.324577 | -0.964125 | -2.705910 | 0.507598 | 0.000000 | -1.735498 | -2.026946 | 1.122145 |
| Epithelial cell | -0.295685 | -1.479849 | 0.841693 | 0.941702 | -2.078219 | -1.604146 | 0.000000 | -0.061404 | -0.451526 | 0.383313 | ... | -0.225143 | -0.583709 | -0.787337 | -0.804225 | 0.040927 | 1.023183 | -2.053631 | 0.081166 | 0.923443 | 0.373183 |
| Macrophage/Monocyte | 0.213761 | 1.048207 | 0.104294 | 3.336899 | 0.309389 | -1.204168 | 0.104333 | -0.442754 | -0.098222 | -0.676066 | ... | -1.014070 | -0.907068 | -0.950240 | -1.287708 | 0.019524 | -0.915923 | -0.413348 | 0.114627 | -0.040516 | -0.169986 |
| Mast cell | -0.512432 | -2.156670 | 0.015692 | -0.621771 | 0.085913 | -1.034468 | 0.000000 | -1.895443 | 1.871222 | 0.000000 | ... | -0.159921 | 1.183988 | -2.084966 | -1.802277 | 1.828191 | -0.903390 | 0.000000 | 0.040864 | 0.819098 | 1.821711 |
| NK cell | -0.172260 | -0.977294 | 0.044326 | -1.355022 | 0.000000 | 1.227618 | 0.000000 | -0.923018 | 0.033855 | 0.000000 | ... | -2.329338 | -1.968455 | -1.900271 | 1.020311 | -0.652086 | -0.963359 | 0.000000 | 0.870265 | -0.119317 | -0.403069 |
| Neutrophils | -0.509581 | 0.949798 | -17.436077 | 0.000000 | -2.722430 | -6.295442 | 0.000000 | 0.655465 | -1.857393 | -0.371984 | ... | -1.509435 | -3.072461 | -2.750386 | 0.000000 | -2.259479 | -4.362793 | -0.050136 | -5.055532 | 0.710715 | -0.988226 |
| Plasma cell | -0.657744 | -0.950014 | 0.473342 | 10.148623 | 0.000000 | -0.374279 | 0.000000 | -0.774725 | 1.027172 | 0.000000 | ... | -2.101581 | 0.730493 | 0.879658 | -0.188601 | -1.784284 | -2.628543 | 0.642968 | 0.346377 | 0.771704 | -0.265788 |
| Stromal | 1.118442 | 0.275912 | 2.433969 | 0.670736 | 1.780228 | -1.413742 | 0.000000 | -0.096848 | -3.396380 | 0.000000 | ... | -1.237317 | 1.343652 | 1.480431 | 2.012742 | -0.747834 | -1.682820 | 0.920978 | -0.127899 | 0.664397 | 1.995964 |
| T cell | -0.002302 | -0.301666 | 1.516062 | 0.933701 | -2.442231 | -2.210447 | 0.000000 | 0.270153 | -0.141248 | 0.000000 | ... | -0.320019 | -0.123477 | -1.222072 | -0.004070 | -0.212104 | -1.275029 | -2.289152 | 1.071218 | -0.127031 | 0.277859 |
| cDC | -0.487030 | -0.749548 | -0.731647 | 30.000000 | 0.000000 | -4.107415 | 1.103510 | -0.434452 | -0.511586 | 0.000000 | ... | -0.775277 | -0.440356 | -1.715100 | -0.648863 | -1.918479 | -1.909194 | 0.000000 | 0.971490 | 0.250980 | 0.182809 |
11 rows × 17835 columns
| gene_id | A1BG | A1BG-AS1 | A2M | A2M-AS1 | A2ML1 | A4GALT | A4GNT | AAAS | AACS | AADAC | ... | ZW10 | ZWILCH | ZWINT | ZXDA | ZXDB | ZXDC | ZYG11A | ZYG11B | ZYX | ZZEF1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cell_type | |||||||||||||||||||||
| B cell | 1.000000 | 1.000000 | 1.000000 | 1.000000e+00 | 1.000000 | 1.000000 | 1.0 | 1.000000 | 1.000000 | 1.000000 | ... | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 |
| Endothelial cell | 0.999824 | 0.999824 | 0.999824 | 9.998236e-01 | 1.000000 | 0.999824 | 1.0 | 0.999824 | 0.999824 | 0.999824 | ... | 0.999824 | 0.999824 | 1.000000 | 0.999824 | 0.999824 | 0.999824 | 1.000000 | 0.999824 | 0.999824 | 0.999824 |
| Epithelial cell | 1.000000 | 1.000000 | 1.000000 | 1.000000e+00 | 1.000000 | 0.895368 | 1.0 | 1.000000 | 1.000000 | 1.000000 | ... | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 |
| Macrophage/Monocyte | 1.000000 | 1.000000 | 1.000000 | 1.000000e+00 | 1.000000 | 1.000000 | 1.0 | 1.000000 | 1.000000 | 1.000000 | ... | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 |
| Mast cell | 1.000000 | 1.000000 | 1.000000 | 1.000000e+00 | 1.000000 | 1.000000 | 1.0 | 1.000000 | 1.000000 | 1.000000 | ... | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 |
| NK cell | 1.000000 | 1.000000 | 1.000000 | 1.000000e+00 | 1.000000 | 1.000000 | 1.0 | 1.000000 | 1.000000 | 1.000000 | ... | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 |
| Neutrophils | 0.997347 | 0.997347 | 0.997347 | 1.000000e+00 | 0.997347 | 0.997347 | 1.0 | 0.997347 | 0.997347 | 0.997347 | ... | 0.997347 | 0.997347 | 0.997347 | 1.000000 | 0.997347 | 0.997347 | 0.997347 | 0.997347 | 0.997347 | 0.997347 |
| Plasma cell | 1.000000 | 1.000000 | 1.000000 | 1.000000e+00 | 1.000000 | 1.000000 | 1.0 | 1.000000 | 1.000000 | 1.000000 | ... | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 |
| Stromal | 1.000000 | 1.000000 | 0.914022 | 1.000000e+00 | 1.000000 | 1.000000 | 1.0 | 1.000000 | 1.000000 | 1.000000 | ... | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 |
| T cell | 1.000000 | 1.000000 | 1.000000 | 1.000000e+00 | 1.000000 | 1.000000 | 1.0 | 1.000000 | 1.000000 | 1.000000 | ... | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 |
| cDC | 1.000000 | 1.000000 | 1.000000 | 2.248608e-09 | 1.000000 | 1.000000 | 1.0 | 1.000000 | 1.000000 | 1.000000 | ... | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 | 1.000000 |
11 rows × 17835 columns
| gene_id | A1BG | A1BG-AS1 | A2M | A2M-AS1 | A2ML1 | A4GALT | A4GNT | AAAS | AACS | AADAC | ... | ZW10 | ZWILCH | ZWINT | ZXDA | ZXDB | ZXDC | ZYG11A | ZYG11B | ZYX | ZZEF1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cell_type | |||||||||||||||||||||
| B cell | -0.383690 | -0.678578 | -0.324745 | 0.000000 | 0.000000 | 0.771001 | 0.000000 | 0.521139 | 0.317013 | 0.000000 | ... | 0.859616 | -0.614895 | 0.934472 | -0.329163 | -0.462775 | -0.335932 | -0.429241 | 0.743194 | -1.013070 | -0.609554 |
| Endothelial cell | 0.202531 | 0.204817 | -0.292430 | -0.375980 | 0.000000 | -0.888596 | 0.000000 | -1.044611 | -0.470339 | -0.626531 | ... | -0.196068 | -1.001738 | -0.046108 | -0.122307 | -0.665466 | 0.081233 | 0.000000 | -0.863614 | -1.387939 | 0.312103 |
| Epithelial cell | -0.281843 | -1.077099 | 0.471101 | 0.326846 | -0.810920 | -1.199858 | 0.000000 | -0.079038 | -0.587255 | 0.272171 | ... | -0.214597 | -0.705854 | -0.692998 | -0.456309 | 0.047344 | 1.030972 | -1.372026 | 0.124328 | 1.095754 | 0.552314 |
| Macrophage/Monocyte | 0.245923 | 0.718931 | 0.113399 | 1.305890 | 0.068347 | -0.558468 | 0.023313 | -0.489239 | -0.073686 | -0.150316 | ... | -0.770577 | -0.850690 | -0.680046 | -0.591796 | 0.008840 | -0.866816 | -0.091754 | 0.189784 | -0.110356 | -0.280560 |
| Mast cell | -0.421836 | -0.804879 | 0.007120 | -0.437713 | 0.018279 | -0.222067 | 0.000000 | -1.284893 | 0.985649 | 0.000000 | ... | -0.081938 | 0.637223 | -0.609719 | -0.385503 | 0.683861 | -0.383447 | 0.000000 | 0.032126 | 0.791249 | 0.944674 |
| NK cell | -0.112471 | -0.422822 | 0.016503 | -0.492033 | 0.000000 | 0.329037 | 0.000000 | -0.494309 | 0.019988 | 0.000000 | ... | -0.840882 | -0.789102 | -0.651747 | 0.317774 | -0.185527 | -0.417218 | 0.000000 | 0.417618 | -0.178463 | -0.393982 |
| Neutrophils | -0.080576 | 0.139580 | -2.789692 | 0.000000 | -0.402170 | -1.283834 | 0.000000 | 0.118391 | -0.364433 | -0.054851 | ... | -0.222850 | -0.498802 | -0.406303 | 0.000000 | -0.333724 | -0.856671 | -0.007389 | -1.124746 | 0.226074 | -0.301051 |
| Plasma cell | -0.951297 | -0.437536 | 0.230770 | 2.234010 | 0.000000 | -0.100974 | 0.000000 | -0.759481 | 0.759228 | 0.000000 | ... | -1.070186 | 0.317855 | 0.359392 | -0.058811 | -0.723286 | -1.255779 | 0.228054 | 0.206527 | 0.502978 | -0.223975 |
| Stromal | 0.533466 | 0.121404 | 2.233977 | 0.199485 | 0.349716 | -1.284819 | 0.000000 | -0.056987 | -1.573771 | 0.000000 | ... | -0.432536 | 0.697406 | 0.440902 | 0.396223 | -0.254302 | -1.120269 | 0.180917 | -0.081941 | 0.768656 | 1.163416 |
| T cell | -0.005062 | -0.324646 | 1.258426 | 0.822308 | -0.591063 | -0.937943 | 0.000000 | 0.310876 | -0.136176 | 0.000000 | ... | -0.236136 | -0.109871 | -0.759348 | -0.002494 | -0.172319 | -1.004597 | -0.519392 | 1.002998 | -0.346977 | 0.465835 |
| cDC | -0.507093 | -0.433977 | -0.636550 | 6.880139 | 0.000000 | -1.315658 | 0.250928 | -0.391445 | -0.275287 | 0.000000 | ... | -0.407788 | -0.297272 | -0.754888 | -0.176891 | -1.156002 | -0.985364 | 0.000000 | 0.675094 | 0.460908 | 0.139605 |
11 rows × 17835 columns
5. Infer pathway activities with consensus#
Run
decouplerconsensus method to infer pathway activities from the DESeq2 result using thePROGENymodels.
We use the obtained gene levelwaldstatistics stored instat.
# Infer pathway activities with consensus
for contrast in contrasts:
print(contrast["name"])
pathway_acts, pathway_pvals = dc.run_consensus(mat=contrast["stat_mat"], net=pwnet)
contrast["pathway_acts"] = pathway_acts
contrast["pathway_pvals"] = pathway_pvals
display(pathway_acts)
LUAD_vs_LUSC
| Androgen | EGFR | Estrogen | Hypoxia | JAK-STAT | MAPK | NFkB | PI3K | TGFb | TNFa | Trail | VEGF | WNT | p53 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B cell | -0.246842 | 1.543935 | 1.558608 | -0.183671 | -2.382583 | 0.810554 | 0.546738 | 0.451384 | -0.143148 | 0.073370 | -0.112629 | 0.952661 | 0.301521 | -0.070714 |
| Endothelial cell | 0.265883 | -1.505292 | -0.321566 | -0.994649 | -0.125569 | -1.122115 | -0.584029 | 0.969558 | 1.325543 | -0.051087 | -0.217974 | -1.351533 | -0.744210 | -1.603530 |
| Epithelial cell | -0.066642 | 0.350913 | 0.609130 | -0.638363 | -2.157498 | 1.037603 | 1.325603 | -0.099008 | 0.982241 | 1.344078 | -0.287075 | -0.267846 | 0.073246 | -1.125983 |
| Macrophage/Monocyte | 0.324897 | 0.053649 | 1.877761 | -0.324897 | -2.601908 | 0.518574 | 1.292100 | -0.084467 | 0.196897 | 0.117583 | -0.141024 | -0.004292 | -0.121309 | -0.324401 |
| Mast cell | 0.454180 | 2.103930 | 0.520413 | -0.245265 | -1.853323 | 0.797029 | 1.094156 | 0.280248 | 0.937557 | 1.111588 | 0.062696 | -0.043590 | 0.334270 | -0.055096 |
| NK cell | 0.212747 | 1.816607 | 0.449081 | -0.603643 | -2.053647 | 0.235018 | -0.369732 | 1.132183 | 1.386292 | -0.611621 | -0.477229 | 0.553257 | -0.223066 | 0.540776 |
| Neutrophils | -0.182056 | 0.331990 | 0.078026 | -1.963086 | -1.363948 | -0.124623 | -1.486473 | -0.260542 | -0.215657 | -0.964152 | 1.542883 | -0.581928 | -0.332444 | -0.716360 |
| Plasma cell | -0.350442 | 0.988855 | 0.659004 | -0.224818 | -1.616886 | 0.544039 | 2.189130 | 0.371615 | 1.108352 | 0.892480 | 0.139872 | 0.120283 | 0.825329 | 0.190010 |
| Stromal | 0.019815 | 0.576191 | -0.330965 | -1.326875 | -1.581439 | 0.551950 | 1.604827 | 1.602388 | -1.025598 | 0.574834 | 0.278758 | 0.456741 | -0.604357 | -0.569790 |
| T cell | 0.173866 | -0.164334 | 2.026978 | -0.180541 | -2.333157 | 0.458294 | -0.072737 | -0.640231 | 0.455387 | -0.622412 | -0.121731 | 0.468706 | -0.874931 | -0.989860 |
| cDC | 0.667159 | 0.707137 | -0.197105 | -1.463723 | -1.242213 | 0.455950 | 1.736791 | 1.035391 | -0.004090 | 1.459614 | 0.176106 | 1.096091 | -0.306651 | 0.045747 |
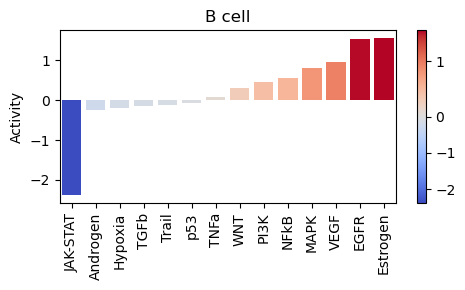
Generate per cell type pathway activity barplots. We use the decoupler
plot_barplotfunction for plotting the pathway activities of each celltype and save the result inpng, pdf, svgformat.
# show maximum n plots in notebook, all are saved as files
show_n = 2
for contrast in contrasts:
print(contrast["name"])
with plt.ioff():
p_count = 0
for ct in contrast["cell_types"]:
bp = dc.plot_barplot(
contrast["pathway_acts"],
ct,
top=25,
vertical=False,
return_fig=True,
figsize=[5, 3],
)
plt.title(ct)
plt.tight_layout()
if bp is not None:
ct_fname = ct.replace(" ", "_").replace("/", "_")
aps.pl.save_fig_mfmt(
bp,
res_dir=f"{contrast['res_dir']}/pathways/",
filename=f"{contrast['name']}_pw_acts_barplot_{ct_fname}",
fmt="all",
)
# Only show the first two plots in the notebook
p_count += 1
if p_count <= show_n:
display(bp)
else:
print(f"Not showing: {ct}")
plt.close()
else:
print("No plot for: " + contrast["name"] + ":" + ct)
print(f"\nResults stored in: {contrast['res_dir']}/pathways")
LUAD_vs_LUSC
Not showing: Epithelial cell
Not showing: Macrophage/Monocyte
Not showing: Mast cell
Not showing: Plasma cell
Not showing: cDC
Not showing: Stromal
Not showing: NK cell
Not showing: Endothelial cell
Not showing: Neutrophils
Results stored in: ../../data/results/10_functional_analysis/LUAD_vs_LUSC/pathways

Generate pathway activity heatmap#
We use the seaborn clustermap function to generate a clustered heatmap of the celltype pathway activities and save the result in png, pdf, svg format.
Signigficant activity differences are marked with “●”
# generate heatmap plot
for contrast in contrasts:
# mark significant activities
sig_mat = np.where(contrast["pathway_pvals"] < 0.05, "●", "")
with plt.rc_context({"figure.figsize": (5.2, 5)}):
chm = sns.clustermap(
contrast["pathway_acts"],
annot=sig_mat,
annot_kws={"fontsize": 12},
center=0,
cmap="bwr",
linewidth=0.5,
cbar_kws={"label": "Pathway activity"},
vmin=-2,
vmax=2,
fmt="s",
figsize=(7, 7),
)
aps.pl.reshape_clustermap(chm, cell_width=0.05, cell_height=0.05)
aps.pl.save_fig_mfmt(
chm,
res_dir=f"{contrast['res_dir']}/pathways/",
filename=f"{contrast['name']}_pw_acts_heatmap",
fmt="all",
)
plt.show()
print(f"\nResults stored in: {contrast['res_dir']}/pathways")

Results stored in: ../../data/results/10_functional_analysis/LUAD_vs_LUSC/pathways
Generate target gene expression plots for significant pathways#
We genereate expression plots for the target genes of pathways with significant activity differences using the DESeq2 wald statistics (y-axis) and the interaction weight (x-axis).
The results are stored in png, pdf, svg format.
Get significant pathways
# filter for p-value < 0.05
for contrast in contrasts:
p = contrast["pathway_pvals"]
sig_pathways_idx = np.where(p < 0.05)
# make list of celltype / pathway pairs
sig_pathways = []
for sig_pw in list(zip(sig_pathways_idx[0], sig_pathways_idx[1])):
ct = p.index[sig_pw[0]]
pw = p.columns[sig_pw[1]]
sig_pathways.append({"ct": ct, "pw": pw})
contrast["sig_pathways"] = sig_pathways
Generate plot
for contrast in contrasts:
print(contrast["name"])
sig_pw = contrast["sig_pathways"]
# Calculate nrows based on ncol
n_sig = len(sig_pw)
ncols = 4 if n_sig >= 4 else n_sig
nrows = int(np.ceil(n_sig / ncols))
# Initialize the figure panel
fig, axs = plt.subplots(nrows=nrows, ncols=ncols, figsize=(ncols * 4, nrows * 4))
empty_axs = axs.flatten()
axs = {": ".join(sig_pw.values()): {"ax": ax, "sig_pw": sig_pw} for sig_pw, ax in zip(sig_pathways, axs.flatten())}
# Run dc.plot_targets for all significant celltype/pathway combinations using the stat values from deseq2
for key, ax_sig_pw in axs.items():
dc.plot_targets(
de_res[ax_sig_pw["sig_pw"]["ct"]].set_index("gene_id"),
stat="stat",
source_name=ax_sig_pw["sig_pw"]["pw"],
net=pwnet,
top=15,
return_fig=False,
ax=ax_sig_pw["ax"],
)
ax_sig_pw["ax"].set_title(key)
# set empty axes invisible
for ax in range(len(axs), len(empty_axs)):
empty_axs[ax].set_visible(False)
plt.tight_layout()
plt.show()
# Save figure
aps.pl.save_fig_mfmt(
fig,
res_dir=f"{contrast['res_dir']}/pathways/",
filename=f"{contrast['name']}_pw_target_expression",
fmt="all",
)
print(f"\nResults stored in: {contrast['res_dir']}/pathways")
LUAD_vs_LUSC
Results stored in: ../../data/results/10_functional_analysis/LUAD_vs_LUSC/pathways

Save pathway activity and p-values matrix#
Finally we store the pathway activity scores and p-values in tsv format.
# save tsv
for contrast in contrasts:
tsv_dir = Path(contrast["res_dir"], "pathways", "tsv")
os.makedirs(tsv_dir, mode=0o750, exist_ok=True)
contrast["pathway_acts"].to_csv(f"{tsv_dir}/{contrast['name']}_pathway_acts.tsv", sep="\t")
contrast["pathway_pvals"].to_csv(f"{tsv_dir}/{contrast['name']}_pathway_pvals.tsv", sep="\t")
6. Infer transcription factor activities with consensus#
Run decoupler consensus method to infer transcription factor activities from the DESeq2 result using the DoRothEA models.
We use the obtained gene level wald statistics stored in stat.
# Infer transcription factor activities with consensus
for contrast in contrasts:
print(contrast["name"])
tf_acts, tf_pvals = dc.run_consensus(mat=contrast["stat_mat"], net=tfnet)
contrast["tf_acts"] = tf_acts
contrast["tf_pvals"] = tf_pvals
display(tf_acts)
LUAD_vs_LUSC
| AHR | AR | ARID2 | ARID3A | ARNT | ASCL1 | ATF1 | ATF2 | ATF3 | ATF4 | ... | ZNF217 | ZNF24 | ZNF263 | ZNF274 | ZNF384 | ZNF584 | ZNF592 | ZNF639 | ZNF644 | ZNF740 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B cell | 0.494807 | -1.295858 | 0.635201 | 1.418887 | -0.102753 | 0.079330 | 0.869259 | 0.407446 | -0.171582 | 0.054380 | ... | 0.229161 | -0.500840 | 1.000174 | 0.400323 | 0.144617 | -0.803723 | 0.255080 | 1.367018 | 0.479102 | 1.965369 |
| Endothelial cell | -0.549029 | -0.115354 | -1.938669 | 0.377054 | -0.778608 | -0.846926 | 0.242634 | -0.825719 | 0.402927 | -0.937720 | ... | -1.901210 | 0.404682 | 1.336712 | -1.310806 | -0.052659 | -0.974487 | -0.098543 | 0.103671 | -0.702017 | -0.527577 |
| Epithelial cell | 0.799596 | 0.889566 | 0.614851 | 0.587870 | -0.160494 | -0.669058 | 1.918827 | 0.276528 | 1.026194 | 0.012645 | ... | 1.058468 | -1.142710 | 0.498509 | 0.835900 | 1.142901 | -0.769006 | -0.369195 | 0.748939 | 1.047694 | -0.379316 |
| Macrophage/Monocyte | 0.658651 | -0.061280 | 1.031650 | 1.948800 | 0.418176 | 0.480713 | 0.321043 | 1.301374 | 0.682867 | 0.072852 | ... | 1.074232 | -0.619390 | 0.412382 | 0.137358 | 0.958108 | -0.207613 | -1.124615 | 1.058357 | 0.958553 | 0.117008 |
| Mast cell | 0.018299 | -0.362015 | 0.351705 | 1.298517 | 0.790628 | -0.003134 | 0.310598 | 2.014740 | 0.951946 | 1.399003 | ... | 0.150157 | 0.076853 | 1.082928 | -0.271137 | 0.011567 | -0.143134 | -0.905225 | 1.381839 | -0.294005 | 0.037592 |
| NK cell | -0.270554 | -0.804563 | 0.147967 | 0.468447 | 0.112974 | 1.337718 | 0.070423 | -0.739791 | -0.257967 | 0.064888 | ... | 1.040004 | 0.472885 | 2.216979 | 0.026001 | 0.443216 | 0.484252 | 0.911763 | 1.514379 | 0.654045 | 2.159696 |
| Neutrophils | 0.042768 | -1.624405 | 0.320883 | -0.345890 | -0.638695 | 0.249693 | -0.880325 | -1.321099 | 0.296990 | -1.317136 | ... | -0.538398 | 0.529491 | 0.347337 | -0.747914 | -1.146214 | -0.443622 | -0.138553 | -0.401604 | 0.452665 | -0.298905 |
| Plasma cell | -0.316239 | -0.512138 | 1.408229 | 0.054453 | 0.374180 | 0.277874 | 0.389478 | 2.105656 | 1.677759 | 2.472244 | ... | -0.287736 | -0.923191 | 0.389007 | -0.030057 | -0.093641 | 0.177626 | 1.005671 | 2.013895 | 0.280040 | 0.210682 |
| Stromal | -0.199434 | -1.809133 | 0.299511 | 0.631933 | 1.150876 | 0.046605 | -0.558228 | -0.047530 | -0.146499 | 1.124004 | ... | -1.005617 | -0.213832 | 1.378833 | 1.708247 | -1.704742 | -0.277866 | -1.160813 | -0.935735 | -0.460369 | -0.501042 |
| T cell | 1.016850 | -1.446534 | 1.498921 | 1.636793 | -0.205554 | -0.167935 | 0.127256 | -0.304119 | 0.396686 | 0.616725 | ... | 0.362580 | 0.122930 | 0.173679 | 0.914766 | 1.057778 | 0.622133 | 1.063043 | 1.867057 | 0.635081 | 1.690971 |
| cDC | 0.952299 | -0.233353 | 0.193726 | -0.543061 | -0.542293 | -0.360546 | 0.758699 | 1.044606 | 0.465229 | 0.867267 | ... | 0.528510 | 0.563882 | 0.865910 | 0.903282 | 0.332664 | 0.016136 | 0.856794 | 0.305656 | 0.244124 | -1.040939 |
11 rows × 297 columns
Generate per cell type transcription factor activity barplots#
We use the decoupler plot_barplot function for plotting the transcription factor activities of each celltype and save the result in png, pdf, svg format.
# show maximum n plots in notebook, all are saved as files
show_n = 2
for contrast in contrasts:
print(contrast["name"])
with plt.ioff():
p_count = 0
for ct in contrast["cell_types"]:
bp = dc.plot_barplot(
contrast["tf_acts"],
ct,
top=25,
vertical=False,
return_fig=True,
figsize=[5, 3],
)
plt.title(ct)
plt.tight_layout()
if bp is not None:
ct_fname = ct.replace(" ", "_").replace("/", "_")
aps.pl.save_fig_mfmt(
bp,
res_dir=f"{contrast['res_dir']}/transcription_factors/",
filename=f"{contrast['name']}_tf_acts_barplot_{ct_fname}",
fmt="all",
)
# Only show the first two plots in the notebook
p_count += 1
if p_count <= show_n:
display(bp)
else:
print(f"Not showing: {ct}")
plt.close()
else:
print("No plot for: " + contrast["name"] + ":" + ct)
print(f"\nResults stored in: {contrast['res_dir']}/transcription_factors")
LUAD_vs_LUSC
Not showing: Epithelial cell
Not showing: Macrophage/Monocyte
Not showing: Mast cell
Not showing: Plasma cell
Not showing: cDC
Not showing: Stromal
Not showing: NK cell
Not showing: Endothelial cell
Not showing: Neutrophils
Results stored in: ../../data/results/10_functional_analysis/LUAD_vs_LUSC/transcription_factors


Generate transcription factor activity heatmap#
We use the seaborn clustermap function to generate a clustered heatmap of the celltype transcription factor activities and save the result in png, pdf, svg format.
Signigficant activity differences are marked with “●”
# generate heatmap plot
for contrast in contrasts:
# get acts and pvals
pvals = contrast["tf_pvals"].copy()
acts = contrast["tf_acts"].copy()
# select the columns that have significant acts (pval < 0.05)
sig_pval_cols = pvals.columns[(pvals < 0.05).any()]
pvals = pvals[sig_pval_cols]
acts = acts[sig_pval_cols]
# mark significant activities
sig_mat = np.where(pvals < 0.05, "●", "")
with plt.rc_context({"figure.figsize": (10, 5)}):
chm = sns.clustermap(
acts,
annot=sig_mat,
annot_kws={"fontsize": 10},
center=0,
cmap="bwr",
linewidth=0.5,
cbar_kws={"label": "TF activity"},
vmin=-2,
vmax=2,
fmt="s",
xticklabels=True,
figsize=(4.5, 4.5),
)
aps.pl.reshape_clustermap(chm, cell_width=0.05, cell_height=0.05)
aps.pl.save_fig_mfmt(
chm,
res_dir=f"{contrast['res_dir']}/transcription_factors/",
filename=f"{contrast['name']}_tf_acts_heatmap",
fmt="all",
)
plt.tight_layout()
plt.show()
print(f"\nResults stored in: {contrast['res_dir']}/transcription_factors")

Results stored in: ../../data/results/10_functional_analysis/LUAD_vs_LUSC/transcription_factors
Volcano plots of expression of target genes from transcription factors of interest#
We genereate volcano plots for the target genes of selected transcription factors with significant activity differences using the DESeq2 log2foldChange (x-axis) and padj (y-axis) values.
For each transcription factor of interest a panel of celltype specific volcano plots will be created. The results are stored in png, pdf, svg format.
tf_of_interest: List of transcription factors in which we are interested and for whose target genes we want to generate a volcano plot
# Define transcription factors of interest
tf_of_interest = ["CEBPA", "SOX13", "SPI1"]
for contrast in contrasts:
# Extract logFCs and pvals
logFCs = contrast["lfc_mat"]
pvals = contrast["fdr_mat"]
tf_pvals = contrast["tf_pvals"]
# get sig ct for tfoi
n_sig = 0
sig_tf = {}
for ct in contrast["cell_types"]:
for tfoi in tf_of_interest:
if tf_pvals.loc[ct][tfoi] < 0.05:
if tfoi not in sig_tf:
sig_tf[tfoi] = []
sig_tf[tfoi].append({"ct": ct, "tf": tfoi})
n_sig += 1
# generate a volcano plot panel for each transcription factor of interest:
# the panels show volcano plots for each celltype in which there is a
# signigicant transcription factor activity
for tf in sig_tf.keys():
n_sig = len(sig_tf[tf])
# Calculate nrows based on ncol
ncols = 4 if n_sig >= 4 else n_sig
nrows = int(np.ceil(n_sig / ncols))
# Initialize the figure panel
fig, axs = plt.subplots(nrows=nrows, ncols=ncols, figsize=(ncols * 4, nrows * 4))
empty_axs = axs.flatten()
axs = [{"ct_tf": ct_tf, "ax": ax} for ct_tf, ax in zip(sig_tf[tf], axs.flatten())]
for ax in axs:
dc.plot_volcano(
logFCs,
pvals,
ax["ct_tf"]["ct"],
name=ax["ct_tf"]["tf"],
net=tfnet,
top=10,
sign_thr=0.1,
lFCs_thr=0.5,
return_fig=False,
ax=ax["ax"],
)
# set empty axes invisible
for ax in range(len(axs), len(empty_axs)):
empty_axs[ax].set_visible(False)
plt.tight_layout()
plt.show()
# Save figure
aps.pl.save_fig_mfmt(
fig,
res_dir=f"{contrast['res_dir']}/transcription_factors/",
filename=f"{contrast['name']}_{tf}_target_expression",
fmt="all",
)
print(f"\nResults stored in: {contrast['res_dir']}/transcription_factors")



Results stored in: ../../data/results/10_functional_analysis/LUAD_vs_LUSC/transcription_factors
Save transcription factor activity and p-values matrix#
Finally we store the transcription factor activity scores and p-values in tsv format.
# save tsv
for contrast in contrasts:
tsv_dir = Path(contrast["res_dir"], "transcription_factors", "tsv")
os.makedirs(tsv_dir, mode=0o750, exist_ok=True)
contrast["tf_acts"].to_csv(f"{tsv_dir}/{contrast['name']}_transcription_factor_acts.tsv", sep="\t")
contrast["tf_pvals"].to_csv(f"{tsv_dir}/{contrast['name']}_transcription_factor_pvals.tsv", sep="\t")
7. Infer enrichment of biological terms with GSEA using significant differential expressed genes#
We can utilize MSigDB to assign biological terms to the differentially expressed genes. In this case, we will employ the run_gsea method from decoupler.
Run GSEA#
We use the wald statistics (stat) from the DESeq2 result to rank the genes.
for contrast in contrasts:
# run_gsea from decoupler
gsea_estimate, gsea_norm, gsea_pvals = dc.run_gsea(
contrast["stat_mat"],
msigdb,
source="geneset",
target="genesymbol",
times=1000,
batch_size=10000,
min_n=5,
seed=4711,
verbose=False,
)
contrast["gsea_estimate"] = gsea_estimate
contrast["gsea_norm"] = gsea_norm
contrast["gsea_pvals"] = gsea_pvals
Now we correct for multiple hypothesis testing using BH
for contrast in contrasts:
# make long format
gsea_pvals = contrast["gsea_pvals"]
gsea_pvals_long = pd.melt(
gsea_pvals.T.rename_axis("source").reset_index(),
id_vars=["source"],
var_name="cell_type",
value_name="pval",
)
# run fdrcorrection and add padj to df
gsea_pvals_long["padj"] = statsmodels.stats.multitest.fdrcorrection(gsea_pvals_long["pval"].values)[1]
# make padj_mat
gsea_padj = gsea_pvals_long.pivot(index="cell_type", columns="source", values="padj")
gsea_padj.index.name = None
# store the padj for the contrast and make sure that we preserve the order of row and columns
contrast["gsea_padj"] = gsea_padj.reindex(index=contrast["gsea_pvals"].index).reindex(
contrast["gsea_pvals"].columns, axis=1
)
Generate MSigDB heatmap from GSEA normalized enrichment scores#
We use the seaborn heatmap function to generate a heatmap of the biological terms assigned by GSEA to the different celltypes and save the result in png, pdf, svg format.
Signigficantly overrepresented terms are marked with “●”
NES (normalized enrichment score) is color-coded
# Plot heatmap of GSEA result
for contrast in contrasts:
gsea_padj = contrast["gsea_padj"]
gsea_norm = contrast["gsea_norm"].copy()
# filter for enriched (positve nes score)
gsea_norm[gsea_norm < 0] = 0
# mark significant enrichment
sig_mat = np.where(gsea_padj[gsea_norm > 0] < 0.05, "●", "")
# create figure
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(17, 10))
# create heatmap
hm = sns.heatmap(
gsea_norm,
annot=sig_mat,
robust=True,
cmap="viridis",
linewidth=0.5,
cbar=True,
cbar_kws={"label": "normalized enrichment score", "shrink": 0.4},
fmt="s",
square=True,
xticklabels=True,
ax=ax,
)
plt.tight_layout()
aps.pl.save_fig_mfmt(
fig,
res_dir=f"{contrast['res_dir']}/MSigDB/",
filename=f"{contrast['name']}_MSigDB_GSEA_heatmap",
fmt="all",
)
plt.show()
print(f"\nResults stored in: {contrast['res_dir']}/MSigDB")

Results stored in: ../../data/results/10_functional_analysis/LUAD_vs_LUSC/MSigDB
Visualize the most enriched terms as barplot#
We use the decoupler plot_barplot function to generate barplots of the most enriched biological and save the result in png, pdf, svg format.
top_n: top n cell types to generate barplots for. Cell types are sorted by the highest GSEA score
top_n = 4
# show top n celltypes
for contrast in contrasts:
print(contrast["name"] + "\n")
display(np.min(contrast["gsea_padj"].T, axis=0).sort_values().head(top_n))
LUAD_vs_LUSC
B cell 9.453365e-14
Neutrophils 6.443057e-11
Epithelial cell 2.340751e-10
Mast cell 5.205902e-09
dtype: float64
for contrast in contrasts:
gsea_norm = contrast["gsea_norm"].copy()
# filter for enriched (positve nes score)
gsea_norm[gsea_norm < 0] = 0
# get top n celltypes
top_celltypes = np.min(gsea_padj.T, axis=0).sort_values().head(top_n).index.values
with plt.rc_context({"figure.figsize": (8, 3)}):
for ct in top_celltypes:
vcenter = np.max(gsea_norm.T[ct]) / 2
bp = dc.plot_barplot(
gsea_norm,
ct,
top=10,
vertical=True,
return_fig=True,
vmin=0,
vcenter=vcenter,
cmap="Reds",
figsize=[8, 3],
)
plt.title(ct)
plt.xlabel("normalized enrichment score")
plt.tight_layout()
plt.show()
if bp is not None:
ct_fname = ct.replace(" ", "_").replace("/", "_")
aps.pl.save_fig_mfmt(
bp,
res_dir=f"{contrast['res_dir']}/MSigDB/",
filename=f"{contrast['name']}_top_terms_barplot_{ct_fname}",
fmt="all",
)
else:
print("No plot for: " + contrast["name"] + ":" + ct)
plt.close()
print(f"\nResults stored in: {contrast['res_dir']}/MSigDB")




Results stored in: ../../data/results/10_functional_analysis/LUAD_vs_LUSC/MSigDB
Generate volcano plots for the most enriched term of the top n celltypes#
top_n: number of celltypes to generate volcano plot of the most enriched term
top_n = 6
# show top n celltypes
for contrast in contrasts:
print(contrast["name"] + "\n")
display(np.min(contrast["gsea_padj"].T, axis=0).sort_values().tail(top_n))
LUAD_vs_LUSC
cDC 1.483697e-07
Plasma cell 5.775455e-07
Stromal 6.139488e-07
Endothelial cell 3.132816e-06
T cell 6.968669e-06
NK cell 3.174736e-05
dtype: float64
for contrast in contrasts:
logFC = contrast["lfc_mat"]
pvals = contrast["fdr_mat"]
# get top n celltypes
top_celltypes = np.min(contrast["gsea_padj"].T, axis=0).sort_values().head(top_n).index.values
# get top term of each celltype
top_term = {}
for ct in top_celltypes:
top_term[ct] = contrast["gsea_norm"].loc[ct].idxmax()
# Calculate nrows based on ncol
ncols = 3 if top_n >= 3 else n_sig
nrows = int(np.ceil(top_n / ncols))
# Initialize the figure panel
fig, axs = plt.subplots(nrows=nrows, ncols=ncols, figsize=(ncols * 6, nrows * 4))
empty_axs = axs.flatten()
axs = [[{"ct": t, "term": top_term[t]}, ax] for t, ax in zip(top_term, axs.flatten())]
for t, ax in axs:
dc.plot_volcano(
logFCs,
pvals,
t["ct"],
name=t["term"],
net=msigdb,
top=10,
sign_thr=0.1,
lFCs_thr=0.5,
source="geneset",
target="genesymbol",
weight=None,
return_fig=False,
ax=ax,
)
# set empty axes invisible
for ax in range(len(axs), len(empty_axs)):
empty_axs[ax].set_visible(False)
plt.tight_layout()
plt.show()
# Save figure
aps.pl.save_fig_mfmt(
fig,
res_dir=f"{contrast['res_dir']}/MSigDB/",
filename=f"{contrast['name']}_top_terms_target_expression",
fmt="all",
)
print(f"\nResults stored in: {contrast['res_dir']}/MSigDB")

Results stored in: ../../data/results/10_functional_analysis/LUAD_vs_LUSC/MSigDB
Save GSEA scores of enriched terms#
Finally we store the GSEA scores of enriched terms in tsv format.
# save tsv
for contrast in contrasts:
tsv_dir = Path(contrast["res_dir"], "MSigDB", "tsv")
os.makedirs(tsv_dir, mode=0o750, exist_ok=True)
contrast["gsea_norm"].to_csv(f"{tsv_dir}/{contrast['name']}_MSigDB_GSEA_nes.tsv", sep="\t")
contrast["gsea_estimate"].to_csv(f"{tsv_dir}/{contrast['name']}_MSigDB_GSEA_estimate.tsv", sep="\t")
contrast["gsea_pvals"].to_csv(f"{tsv_dir}/{contrast['name']}_MSigDB_GSEA_pvals.tsv", sep="\t")
contrast["gsea_padj"].to_csv(f"{tsv_dir}/{contrast['name']}_MSigDB_GSEA_padj.tsv", sep="\t")
8. CytoSig analysis#
We define enriched cytokine signaling signatures in the tumor cells using the CytoSig signature matrix an the decoupler consesus scoring function.
First we reformat the signature matrix into long format
# reformat the signature matrix
cyto_sig = pd.melt(
cytosig_signature.rename_axis("target").reset_index(),
var_name="source",
id_vars=["target"],
value_name="weight",
).reindex(columns=["source", "target", "weight"])
cyto_sig
| source | target | weight | |
|---|---|---|---|
| 0 | Activin A | A2M | 0.050964 |
| 1 | Activin A | A4GALT | -0.208403 |
| 2 | Activin A | AAAS | -0.026028 |
| 3 | Activin A | AACS | -0.107110 |
| 4 | Activin A | AAK1 | -0.022077 |
| ... | ... | ... | ... |
| 329761 | WNT3A | ZZZ3 | 0.027164 |
| 329762 | WNT3A | CHN2 | -0.064087 |
| 329763 | WNT3A | ESRRG | 0.042413 |
| 329764 | WNT3A | FHIT | -0.046138 |
| 329765 | WNT3A | HOXB6 | 0.053906 |
329766 rows × 3 columns
Run the decoupler consensus scoring function using the DESeq2 wald statistics and the CytoSig net
# Infer cytokin signaling with consensus
for contrast in contrasts:
print(contrast["name"])
cs_acts, cs_pvals = dc.run_consensus(mat=contrast["stat_mat"], net=cyto_sig)
contrast["cs_acts"] = cs_acts
contrast["cs_pvals"] = cs_pvals
display(cs_acts)
LUAD_vs_LUSC
| Activin A | BDNF | BMP2 | BMP4 | BMP6 | CD40L | CXCL12 | EGF | FGF2 | GCSF | ... | PGE2 | TGFA | TGFB1 | TGFB2 | TGFB3 | TNFA | TRAIL | TWEAK | VEGFA | WNT3A | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B cell | -0.482646 | 2.259695 | -0.863616 | -1.478322 | 0.045383 | -0.587955 | -1.131240 | 0.708744 | -2.260390 | 0.762260 | ... | 0.096722 | 0.382647 | -0.597530 | 1.654242 | -1.672500 | 0.085604 | -0.374362 | -0.116501 | 0.441778 | 0.904713 |
| Endothelial cell | -0.140653 | -1.391896 | 0.178591 | 0.526312 | 1.052298 | 0.599148 | -0.009525 | -1.536898 | 0.367362 | 0.183483 | ... | -0.882414 | -0.814086 | 0.080817 | -0.311397 | 0.818801 | 0.118420 | 0.831849 | -0.907669 | -0.890997 | 1.998671 |
| Epithelial cell | 0.272666 | 0.784764 | 0.310924 | 1.235774 | -0.914101 | 1.092303 | 0.749393 | 0.998541 | 0.405041 | 0.557434 | ... | 1.420431 | 0.623688 | 1.169039 | 0.339757 | 2.496500 | 0.739314 | 0.873977 | 0.676544 | 0.679720 | 0.279122 |
| Macrophage/Monocyte | 0.041470 | 0.338627 | 0.317325 | 0.641722 | 0.309349 | 1.157407 | 0.188017 | 1.147379 | 0.363295 | 1.282793 | ... | 0.391389 | 0.514752 | 0.716912 | 0.610758 | 1.851552 | 0.948503 | 0.728291 | 0.287427 | 1.120714 | 0.509945 |
| Mast cell | -0.096104 | 0.772115 | -0.267422 | 0.273004 | -0.017583 | 1.223186 | 0.569611 | 1.404026 | 0.896434 | 0.931618 | ... | 0.340526 | 1.117030 | 0.683753 | 0.585125 | 0.333965 | 0.926995 | 1.497205 | -0.190361 | 2.420311 | -0.224754 |
| NK cell | 0.579675 | 1.941291 | 0.323327 | -0.651686 | 0.393145 | 0.514874 | 0.541557 | 0.720228 | -1.358832 | 0.430220 | ... | 1.045958 | 0.888556 | 0.256206 | 0.933542 | 1.715606 | 1.100328 | 0.643063 | 1.301733 | 0.427909 | 0.206403 |
| Neutrophils | -0.142243 | -0.284395 | -0.146106 | 0.043163 | 1.370602 | 0.466852 | -0.735402 | -1.045701 | 0.103361 | 0.851347 | ... | 0.029674 | -0.742348 | 0.028969 | -0.268258 | 0.710881 | -0.522137 | 1.238972 | -0.554146 | -0.016647 | 1.303866 |
| Plasma cell | 0.256896 | 0.878637 | 0.040202 | -0.118374 | -0.959249 | 0.492737 | 0.818973 | 1.345826 | -0.165030 | 0.694331 | ... | 0.916952 | 1.382699 | 0.226926 | 1.535075 | 1.177693 | 1.292434 | 0.997565 | 0.339793 | 1.494568 | 0.082868 |
| Stromal | -0.200350 | -0.317472 | -0.636725 | -1.060266 | 0.785712 | 1.281654 | 0.419830 | -0.099255 | 0.321945 | 0.117448 | ... | 0.444467 | -1.360930 | -0.758919 | -1.296940 | -0.953828 | 0.372752 | 2.755995 | -0.088483 | -0.576638 | -0.388535 |
| T cell | 0.140575 | 1.639995 | -0.324083 | -0.616470 | -0.592388 | 0.268146 | -0.642872 | 1.222686 | -0.956480 | -0.371912 | ... | 0.765180 | 0.291763 | 0.594390 | 1.611180 | 1.894272 | 0.106447 | 0.613308 | 0.675075 | 0.567940 | 0.762486 |
| cDC | -0.167750 | -0.032822 | 0.477761 | 0.674290 | 0.665356 | 2.155193 | 0.137226 | 0.423278 | -0.292649 | 0.860476 | ... | 0.159508 | 0.462986 | 0.766819 | 0.040337 | 1.995206 | 1.628578 | 1.295601 | 0.865028 | 0.308939 | 0.840638 |
11 rows × 51 columns
Generate signaling activity heatmap#
We use the seaborn clustermap function to generate a clustered heatmap of the celltype signaling activities and save the result in png, pdf, svg format.
Signigficant signaling activity differences are marked with “●”
# generate heatmap plot
for contrast in contrasts:
# get acts and pvals
pvals = contrast["cs_pvals"].copy()
acts = contrast["cs_acts"].copy()
# select the columns that have significant acts (pval < 0.05)
sig_pval_cols = pvals.columns[(pvals < 0.05).any()]
pvals = pvals[sig_pval_cols]
acts = acts[sig_pval_cols]
# mark significant activities
sig_mat = np.where(pvals < 0.05, "●", "")
with plt.rc_context({"figure.figsize": (10, 10)}):
chm = sns.clustermap(
acts,
annot=sig_mat,
annot_kws={"fontsize": 10},
center=0,
cmap="bwr",
linewidth=0.5,
cbar_kws={"label": "signaling activity"},
vmin=-2,
vmax=2,
fmt="s",
xticklabels=True,
figsize=(6, 6),
)
aps.pl.reshape_clustermap(chm, cell_width=0.05, cell_height=0.05)
aps.pl.save_fig_mfmt(
chm,
res_dir=f"{contrast['res_dir']}/cytokine_signaling/",
filename=f"{contrast['name']}_signaling_heatmap",
fmt="all",
)
plt.show()
print(f"\nResults stored in: {contrast['res_dir']}/cytokine_signaling")

Results stored in: ../../data/results/10_functional_analysis/LUAD_vs_LUSC/cytokine_signaling
Save cytokine signaling activity scores and p-values matrix#
Finally we store the cytokine signaling activity scores and p-values in tsv format.
# save tsv
for contrast in contrasts:
tsv_dir = Path(contrast["res_dir"], "cytokine_signaling", "tsv")
os.makedirs(tsv_dir, mode=0o750, exist_ok=True)
contrast["cs_acts"].to_csv(f"{tsv_dir}/{contrast['name']}_CytoSig_acts.tsv", sep="\t")
contrast["cs_pvals"].to_csv(f"{tsv_dir}/{contrast['name']}_CytoSig_pvals.tsv", sep="\t")